
Introduction
Bringing a new drug to market costs an average of $2.56 billion and takes 10–15 years, according to PhRMA. Despite that investment, only 12% of new molecular entities entering clinical trials ever receive FDA approval. The Phase II success rate sits at a sobering 28.9%.
Those numbers represent more than R&D inefficiency. Behind each failed compound are patients waiting years for treatments that never arrive, and capital that could have funded multiple programs spent on a single late-stage collapse.
Machine learning is changing how researchers approach each stage of this process — from identifying biological targets to designing candidate molecules computationally to running smarter clinical trials.
This article covers where ML is delivering real results across the drug discovery pipeline, the manufacturing floor, and regulatory workflows, plus an honest look at the challenges still standing in the way.
Key Takeaways
- ~90% of drug candidates fail in clinical trials — ML tools can flag efficacy and toxicity risks before trials begin
- Generative AI models design novel molecules from scratch rather than screening existing compound libraries
- Patient biomarker stratification nearly doubles clinical approval probability vs. unselected populations
- AlphaFold resolved a 50-year protein structure prediction challenge, compressing timelines that once took years into hours
- Insilico Medicine's AI-designed compound completed discovery-to-candidate selection in 18 months
- Fragmented data infrastructure remains the primary barrier to ML adoption — not model quality
Why Drug Discovery Desperately Needs Machine Learning
The Eroom's Law Problem
Pharma R&D has a paradox at its core. As technology has advanced, drug development has gotten harder, not easier. Scannell et al.'s landmark 2012 analysis in Nature Reviews Drug Discovery documented this as Eroom's Law: the number of new drugs approved per $1 billion of R&D spending has halved roughly every 9 years since 1950, representing an 80-fold decline in inflation-adjusted productivity. PhRMA member companies have invested more than $1 trillion in R&D since 2000 with that declining return.
Incremental process improvements (faster lab equipment, better assays, more efficient clinical operations) haven't reversed this trend. The issue isn't execution — it's that the underlying process of identifying the right targets and compounds generates too many costly late-stage surprises.
Three Failure Modes Driving the Attrition Rate
Research published in Acta Pharmaceutica Sinica B breaks down why roughly 90% of clinical drug development fails:
- Lack of efficacy — 40–50% of clinical failures; often traced back to pursuing targets not actually driving disease
- Unmanageable toxicity — ~30% of failures; frequently discovered only in late-stage trials when costs are highest
- Poor drug-like properties — 10–15% of failures; ADMET-related issues that early-stage screening missed
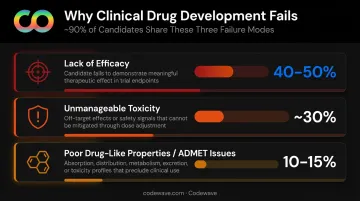
Late-stage discovery of any of these problems is catastrophic. A Phase III failure can erase hundreds of millions in sunk costs with nothing to show for it.
Why ML Fits This Problem
Traditional drug discovery is sequential and lab-heavy. Each hypothesis requires physical synthesis and testing before the next step. ML inverts this by:
- Scanning millions of compounds or data points computationally before any synthesis occurs
- Learning from historical failure patterns to predict which candidates will fail and why
- Improving predictions continuously as more experimental data is generated
In practice, this means pharma teams can screen candidate libraries orders of magnitude larger than wet lab throughput allows — then enter the lab with a shortlist ranked by predicted efficacy and safety, not just chemical availability. Early ML-assisted screening programs have reported reducing compound libraries from millions to hundreds of prioritized candidates before a single synthesis run.
Key ML Applications Across the Drug Discovery Pipeline
Target Identification and Validation
Finding the right biological target — the protein, gene, or pathway implicated in a disease — is where most drug programs succeed or fail before a molecule is ever designed.
ML, particularly natural language processing and knowledge graph models, mines genomic databases, proteomic data, and decades of biomedical literature simultaneously. BenevolentAI's knowledge graph, for example, contains more than 350 million biomedically relevant relationships extracted from scientific literature, enabling the system to surface target-disease connections that human researchers would take years to identify manually.
For rare diseases — where more than 90% still lack effective therapies and patient datasets are scarce — this computational reach is especially valuable.
ML models can simulate target-compound interactions to validate or rule out candidates before any wet-lab work begins, compressing years of exploratory biology into weeks of computation.
Generative Molecule Design and Virtual Screening
Traditional high-throughput screening tests whether existing compounds bind to a target. Generative AI designs new molecular structures from scratch, optimized for specific properties rather than selected from existing libraries.
Generative model architectures such as Variational Autoencoders (VAEs) and Generative Adversarial Networks (GANs) produce novel molecules with tunable characteristics — binding affinity, metabolic stability, low toxicity — that don't exist in any compound library. In a 2019 Nature Biotechnology study, researchers used a generative model (GENTRL) to identify potent DDR1 kinase inhibitors in just 21 days, including synthesis and testing.
Once candidate molecules are generated, virtual screening ranks them by predicted activity and safety — before a single compound is physically synthesized:
- Validated virtual screening methods produce hit rates ranging from 1% to 40%, dramatically narrowing the candidate field
- ML-assisted hit prioritization for high-throughput screening has been validated in ACS Central Science research
- Fewer compounds synthesized means faster timelines and lower costs
ADMET Prediction and Toxicity Screening
ADMET properties — Absorption, Distribution, Metabolism, Excretion, Toxicity — determine real-world drug viability, and failures here account for up to 45% of late-stage clinical failures.
ML models trained on historical compound data can flag ADMET problems early, when course corrections are cheap. The ADMET-AI platform, trained on 41 ADMET datasets, demonstrates what this looks like at scale:
- AUROC > 0.85 on 20 of 31 classification tasks
- Screens 1 million molecules in 3.1 hours using standard computing hardware
- 45% faster than the next-fastest public ADMET web server

Catching a toxicity problem at the computational stage — rather than in Phase II trials — can save years of development time and tens of millions in sunk costs.
How ML Is Reshaping Clinical Trials
Clinical trials are where most drug programs actually die. Around 80% fail to meet initial enrollment targets and timelines, with delays costing drug developers as much as $8 million per day in lost revenue. ML is attacking several of these failure modes simultaneously.
Patient Recruitment and Stratification
Finding eligible patients is harder than it sounds. Eligibility criteria are complex, and patients may never have been formally diagnosed under the exact protocol terminology. Clinical sites rarely have the resources to screen charts manually at scale.
ML algorithms scan de-identified electronic health records and genomic databases to identify patients who match complex eligibility criteria. One validated ensemble ML algorithm reduced the volume of ineligible patients requiring manual chart review by 40–57% while retaining nearly 100% of genuinely eligible candidates — saving an estimated 588 working hours at a single tertiary center.
That efficiency gain matters less if the wrong patients are enrolled. Stratification — specifically biomarker-based preselection — is what ultimately drives approval odds. BIO's 2021 analysis of 9,704 development programs found:
- FDA approval likelihood: 15.9% (with biomarker stratification) vs. 7.6% (without)
- Phase II transition success: 46.3% vs. 28.3%
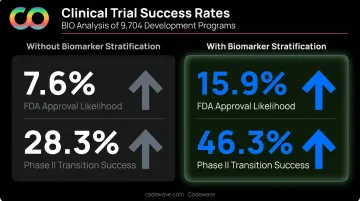
The gap widens at earlier stages, where enrollment decisions have the most downstream impact.
Synthetic Control Arms and Risk-Based Monitoring
Recruitment and stratification address who enters a trial. Two other ML applications change how trials are run:
Synthetic control arms (SCAs) use historical trial data and real-world evidence to construct virtual comparator cohorts, reducing the number of patients assigned to placebo. In rare diseases and oncology — where placebo assignment raises serious ethical concerns — SCAs enable trials that might otherwise be unfeasible. Unlearn's PROCOVA methodology has received FDA commentary, indicating the FDA is open to this methodology in submission dossiers.
Risk-based monitoring (RBM) uses ML to detect data quality issues, protocol deviations, and site performance problems in real time, replacing the traditional model of periodic manual on-site reviews. ACRO white paper analysis found that centralized monitoring approaches produced 28% fewer critical and major findings per quality control visit and 16% lower mean cost per monitoring visit compared to conventional on-site methods.
Real-World Breakthroughs: ML in Action
AlphaFold: Solving a 50-Year Problem
Protein structure determines function. For 50 years, predicting a protein's 3D shape from its amino acid sequence alone was considered one of biology's hardest unsolved problems. DeepMind's AlphaFold solved it.
The database now contains more than 200 million protein structure predictions, covering nearly all catalogued proteins known to science. Researchers can examine the precise shape of a disease-relevant target, including proteins involved in misfolding diseases like Alzheimer's and Parkinson's, without years of crystallography work.
Target identification that once required a full research program can now start from a structure that's already computed.
Insilico Medicine's Rentosertib
The clearest proof that end-to-end AI drug discovery works at clinical scale is Insilico Medicine's rentosertib, a TNIK inhibitor developed for idiopathic pulmonary fibrosis (IPF).
The timeline tells the story:
- ML identified TNIK as the relevant target using multi-omic and literature data
- A generative chemistry engine designed the molecule
- Discovery through preclinical candidate nomination: 18 months, fewer than 80 molecules synthesized
- Phase IIa results published in Nature Medicine in 2025
- Phase III trial initiated in July 2026, enrolling 320 patients across 47 centers
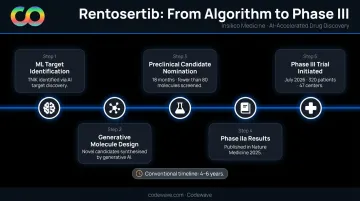
A conventional discovery program for the same indication would typically take 4–6 years to reach the same stage.
MELLODDY: Federated Learning Across Competitors
Drug discovery data is inherently competitive. No pharma company shares its compound libraries. MELLODDY — a consortium of 10 major companies including AstraZeneca, Novartis, GSK, and Merck KGaA — solved this through federated learning.
Rather than pooling raw compound data, the algorithm traveled to each company's data and trained locally, then aggregated model improvements without exposing proprietary structures. Key outcomes:
- All 10 participants saw improved predictive accuracy on their own classification and regression tasks
- Zero proprietary structures were shared across competing organizations
- Every company's internal models benefited from the collective signal
Novo Nordisk: Accelerating Regulatory Documentation
Regulatory submission documents, especially clinical study reports, are among the most time-intensive in pharma. Novo Nordisk's NovoScribe generative AI tool targets this bottleneck directly. Their 2024 Capital Markets Day materials report approximately 70% faster document production for clinical study reports. That speed difference can compress a submission timeline by weeks — meaningful when regulatory queues already stretch months.
Key Challenges to ML Adoption in Pharma
Data Fragmentation
The most common reason ML pilots fail in pharma isn't model architecture — it's data infrastructure. Clinical, preclinical, manufacturing, and real-world data typically sit in separate systems with incompatible formats, inconsistent labeling, and no unified access layer. A model is only as good as the data it's trained on.
Building ML-ready data infrastructure means consolidating these systems, standardizing schemas, and creating automated pipelines that keep training data current. For regulated pharma environments, that work also needs to account for compliance requirements around data handling and audit trails — which adds complexity that many organizations underestimate.
Regulatory and Explainability Requirements
Solving the data problem is only part of the equation. Clean, consolidated data still needs to pass through a regulatory lens before it informs any drug development decision.
FDA and EMA frameworks are evolving quickly. In January 2026, both agencies jointly published "Guiding Principles of Good AI Practice in Drug Development", covering requirements around human-centric design, transparency, risk-based validation, and lifecycle monitoring.
In practice, this means:
- ML models used in drug development must be validated and monitored for drift over time
- High-stakes applications — such as models that inform clinical decisions — require interpretability, not just accuracy
- Model selection and documentation practices must account for what regulators will accept, not just what performs best on benchmarks

Black-box deep learning models may deliver better predictive performance but face higher regulatory scrutiny than simpler, explainable alternatives.
Talent and Organizational Gaps
ML in drug discovery requires people who understand both biology and data science at a working level — a combination that is hard to find. McKinsey's 2025 life sciences analysis identifies talent realignment as one of the primary barriers to scaling AI in the sector.
Beyond individual expertise, organizational culture matters. Many pharma companies have deep institutional practices built around wet-lab experimentation. Shifting workflows to incorporate computational predictions, and getting scientists to trust and act on model outputs, requires change management as much as technical implementation.
Frequently Asked Questions
How does machine learning differ from traditional drug discovery methods?
Traditional discovery relies on sequential physical experimentation — synthesize a compound, test it, repeat. ML uses pattern recognition across large datasets to predict outcomes computationally before experiments run — identifying promising candidates and failure risks faster, at a fraction of the cost.
What types of machine learning are most commonly used in pharma?
Pharma teams rely on three core approaches: supervised learning for predicting outcomes like toxicity or binding affinity; unsupervised learning for clustering patient subgroups or discovering molecular patterns; and deep learning for complex tasks like protein structure prediction and generative molecule design.
What data is needed to apply machine learning in drug discovery?
ML models draw on genomic datasets, compound libraries with known activity and toxicity profiles, electronic health records, clinical trial results, and biomedical literature. Data quality matters more than volume — inconsistently labeled or incomplete data produces unreliable models.
How is ML being used to improve clinical trial success rates?
ML improves trial outcomes through four levers:
- Optimizing patient recruitment using EHR screening
- Stratifying populations via biomarker profiles to sharpen efficacy signals
- Constructing synthetic control arms to reduce placebo assignment
- Monitoring trials in real time to catch protocol issues early
What are the biggest challenges in using ML for drug discovery?
The biggest obstacles are fragmented, low-quality data across siloed systems; difficulty validating and explaining ML models to regulators under evolving FDA/EMA frameworks; and a shortage of professionals with the hybrid expertise needed to bridge biology and data science.
Is machine learning replacing scientists in drug discovery?
No. ML handles pattern recognition and prediction at scale — tasks humans cannot do across millions of data points simultaneously. Scientists remain essential for hypothesis formation, experimental design, and interpreting whether computationally predicted results make biological and clinical sense.


